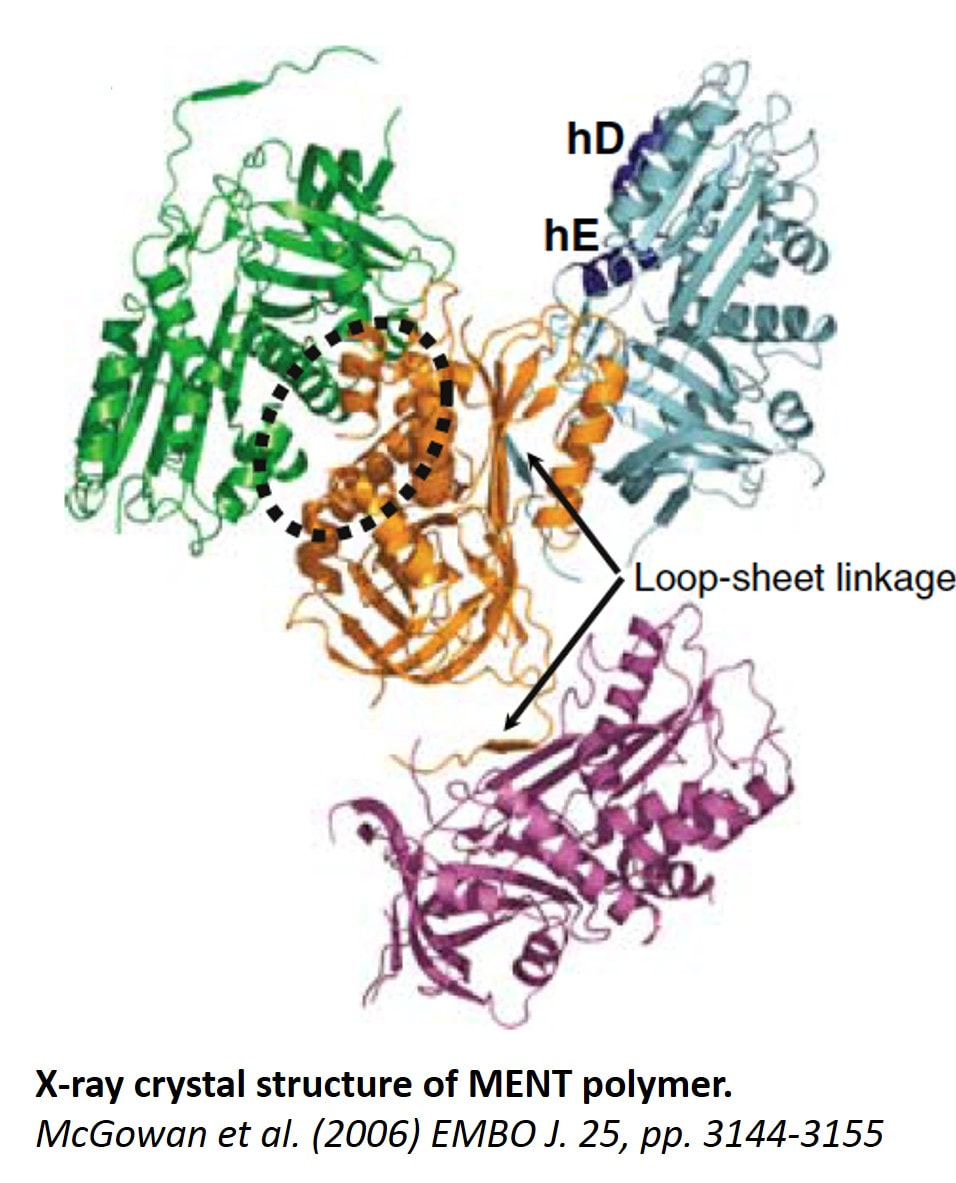
Molecular and structural biology of chromatin architectural factorsWe studied architectural chromatin factors, including chromatin-associated protease inhibitors (serpins), and their role in regulating chromatin epigenetics during cell differentiation and cancer. Our work on the first chromatin-associated serpin, MENT, that I have identified and cloned opened a whole new avenue in structural analysis of chromatin factors associated with nucleosome arrays as MENT was found, by combination of biochemical experiments, electron microscopy, and X-ray crystallography to be the first known chromatin bridging factor and the first nonhistone chromatin factor with solved X-ray structure. A central finding for understanding of chromatin condensation mechanism was its capability to polymerize induced by binding to the nucleosomes (Istomina et al., 2003, Mol Cell Biol 23, 6455-6468; Springhetti et al., 2003, J Biol Chem 278, 43384-43393; McGowan et al., 2006, Embo J 25, 3144-3155). MENT protease inhibition activity lead us to a surprising discovery of unexpected role of lysosomal protease, cathepsin L, in regulating chromatin epigenetics (Bulynko et al., 2006, Mol Cell Biol 26, 4172-4184).
Epigenetic mechanisms underlying cell differentiation and cancer
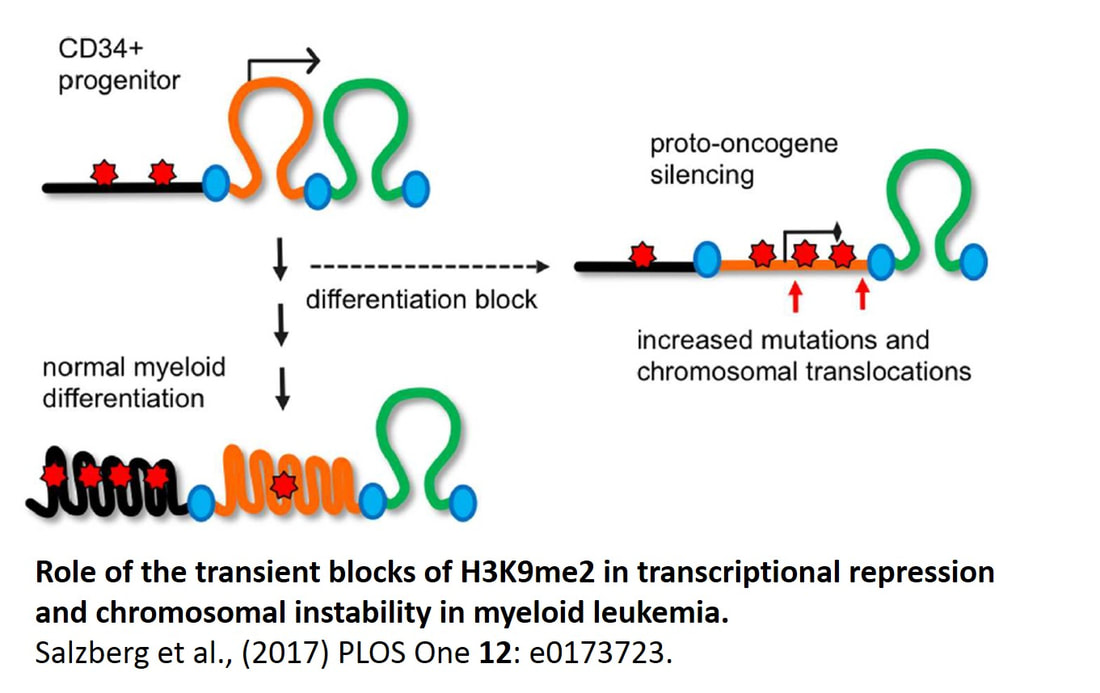
Our main contribution to the field is the discovery that histone H3 methylation at lysine 9, a mark of facultative heterochromatin, plays a major role in 3D spatial organization of chromatin in differentiated cells and its large-scale reorganization is associated with a transition from normal cell to cancer. In particular, we identified a new (apocentric) heterochromatin compartment formed by histone H3 lysine 9 methylation specifically studied in lymphocytes (Grigoryev et al., 2004, J Cell Sci 117, 6153-6162) erythroid cells (Popova et al., 2009, Chromosome Res 17, 47-64) but absent from undifferentiated cells and myeloid leukemia (Popova et al., 2006, Exp Hematol 34, 453-462). This work was spearheaded by a postdoctoral researcher in my lab, E.Popova, who is now working as a research associate at the Department of Neuroscience and continues this line of research in a collaborative study with her and Dr. Colin Barnstable on chromatin structure in developing retina cells (Popova et al., 2013, J. Biol Chem 288, 17895-17907). Since we first observed strong changes in H3 lysine 9 methylation in cancer, we undertook genome-wide mapping of this modification in normal and leukemia myeloid cells in collaboration with Dr. D.Claxton (Penn State Cancer institute) and A.Salzberg (Penn State Institute for Personalized medicine). Our results showed that genome-wide epigenetic changes are abundant in AML and orchestrate illegitimate gene expression in AML cells (Salzberg et al., in preparation). This work leads to a new direction in the cancer epigenetic field that will be actively pursued in my lab in the coming years.
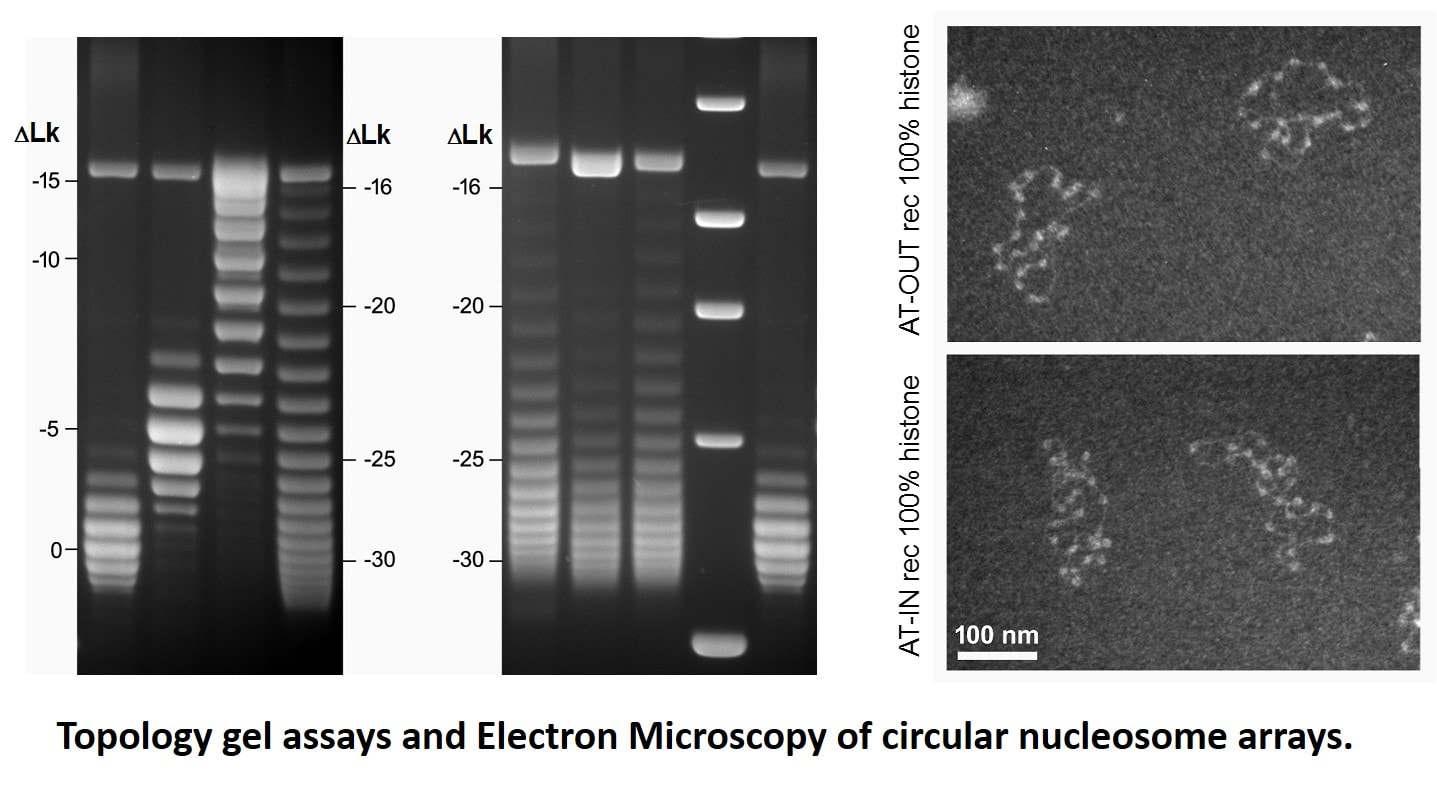
DNA topology in circular nucleosome chainsIn eukaryotic chromatin, the DNA double helix is repeatedly supercoiled making ~1.7left superhelical turns in each nucleosome. However, there is a long-standing discrepancy between the nucleosome structure and measurements of DNA topology in circular nucleosome chains, indicating that there is only ~1.0 superhelical turn per nucleosome. This discrepancy noted by Francis Crick in 1975, is known as the superhelical paradox. Since then, a number of theoretical assumptions were put forward to explain this paradox but none has been tested experimentally. Previously we and others hypothesized that that the geometry of linker DNA could affect topological properties of circular nucleosome chains. To test this hypothesis, we constructed several nucleosome templates with different linker DNA length capable of changing rotational orientation of the nucleosomes. Recently, in collaboration with Dr. Zhurkin and his colleagues at NIH, we analyzed topological properties of DNA in circular nucleosome arrays with precisely positioned nucleosomes.
Using topological electrophoretic assays and electron microscopy, we demonstrated that the number of superhelical turns per nucleosome strongly depends on the nucleosome spacing and varies by as much as 0.5 superhelical turns per nucleosome. With this result, we could reconcile the topological properties of nucleosome arrays with nucleosome core structure and provide a simple explanation for the DNA topology in native chromatin with variable DNA linker length. We also proposed that the newly discovered topological polymorphism of the chromatin fibers may reflect a more general tendency of chromosomal domains, which are organized as topologically separated loops, to maintain topologically distinct higher-order structures and confer different modes on nucleosome interactions within the transcriptionally active and repressed chromatin (Nikitina et al., 2017, Science Adv. 3(10):e1700957). This work was funded by National Science Foundation, award #1516999. |

Chromatin higher-order structure
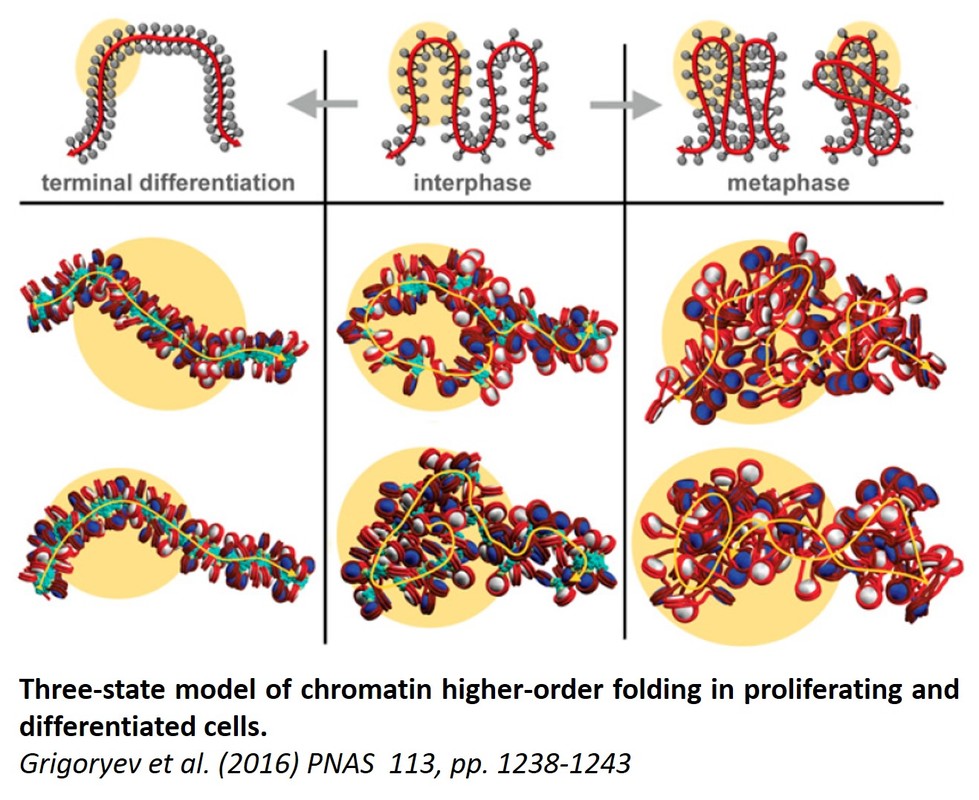
Understanding how chromatin higher-order folding regulates gene accessibility to transcription, replication, and repair is currently a major goal of biomedical research. Since I became an independent NIH-funded investigator in year 1995, I took a leading role in the research effort that eventually changed the previous paradigm of static chromatin solenoidal coiling and lead to appreciation of the flexible and dynamic nucleosome zigzag folding. Our new electron microscopy based technique, EMANIC (EM-assisted nucleosome interaction capture)(Grigoryev et al., 2009, Proc Natl Acad Sci U S A 106, 13317-322) brought new insights in the field by showing that the zigzag and the solenoidal topologies can be simultaneously present in one structurally heteromorphic but energetically stable chromatin fiber. This work reconciles a long-standing controversy (also reflected in a number of recent textbooks) and also opens way to understanding the structural basis of the surprising chromatin accessibility to many DNA-dependent processes. Our recent work on the effect of nucleosome linker length published in EMBO J. (Correll et al., 2012, The EMBO journal 31, 2416-2426) showed that linker length heterogeneity is a major factor of the chromatin fiber structural variations. Most recently, we applied EMANIC to chromatin folding in situ in combination with mesoscopic computational modeling of chromatin fibers by T. Schlick group at NYU. We were able to resolve for the first time the changes in chromatin higher-order folding associated with metaphase chromosome condensation and to propose a three-state model of chromatin folding (Grigoryev et al., 2016, Proc Natl Acad Sci U S A 113, 1238-1243). Furthermore, our advance in understanding the basic principles of chromatin structure has a direct application to biomedical problems. For example, our collaborative papers demonstrate that the higher order structural transitions underlie extracellular chromatin unfolding by arginine citrullination that is involved in bacterial trapping by neutrophils and acute inflammation (Wang et al., 2009, J Cell Biol 184, 205-213) as well as chromatin regulation by NSBP1 (Rochman et al., 2009, Mol Cell 35, 642-656), a chromatin protein associated with human cancer. These studies uncover candidate factors regulating higher-order chromatin structure in vivo that may be targeted by specific drugs and provide new directions for future expansion of our chromatin studies. This work was funded by National Science Foundation, awards # MCB-0615536 and MCB-1021681 .
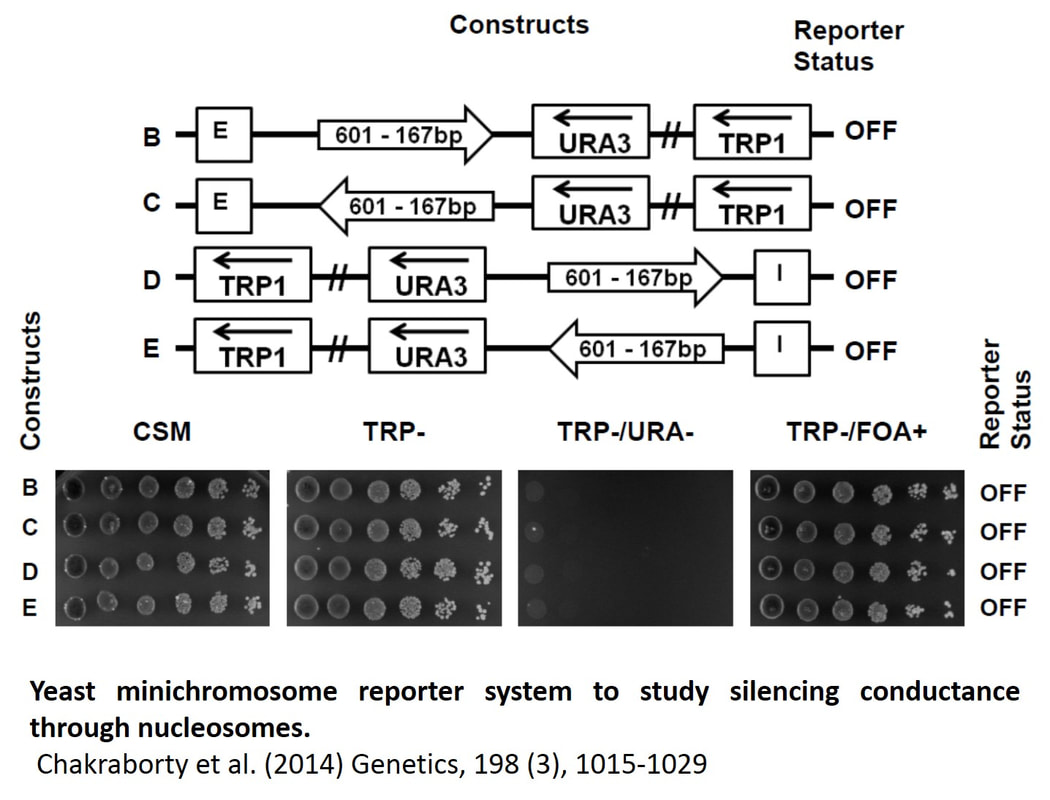
Gene regulation in circular minichromosomesWe have successfully established a yeast minichromosome reporter system in which we were able to reproduce a competition between heterochromatin spreading and chromatin boundaries for gene repression (Chakraborty et al., 2011, PLoS One 6, e24835) Using this system we were able to establish silencing conductance through a string of positioned nucleosomes and determine the nucleosome repeat number limiting silencing (Chakraborty et al., 2014, Genetics 198, 1015-1029). This work opens a new way to study chromatin silencing and associated chromatin structural transitions in a minichromosome system with topologically constrained chromatin.
|